
 当前位置:
当前位置:




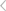

量子化学过渡态模拟计算是一种利用量子化学理论和计算方法,对化学反应过程中过渡态的结构、能量以及反应路径进行模拟预测的技术。过渡态是化学反应路径中的关键步骤,它连接着反应物和产物,并决定了反应的速率和机理。以下是对量子化学过渡态模拟计算的详细阐述:
一、过渡态的基本概念
过渡态是指化学反应过程中,反应物转化为产物时经历的一个高能量、不稳定的中间状态。在势能面上,过渡态位于反应路径上的最高点,也被称为鞍点。通过计算过渡态的结构和能量,可以揭示化学反应的微观机理,并预测反应速率和产物分布。
二、量子化学过渡态模拟计算的方法
-
势能面扫描法:通过改变反应坐标(如键长、键角等),在势能面上进行扫描,找到能量最高的点即为过渡态。这种方法需要预设反应路径,且计算量较大。
-
优化法:利用量子化学计算软件,通过优化算法(如梯度下降法、共轭梯度法等)在势能面上寻找能量最低点,即稳定构型。然后,通过逐步改变结构参数,使系统逐渐接近过渡态,并最终找到能量最高点。这种方法不需要预设反应路径,但需要对初始结构进行合理猜测。
-
过渡态搜索法:如NEB(Nudged Elastic Band)方法、Dimer方法等,这些方法通过构建一系列中间结构(即“图像”),并在这些结构之间施加力或约束,使系统沿着反应路径向过渡态逼近。NEB方法通过在反应路径上撒点,并在点之间施加弹簧力来模拟过渡态的搜索过程;Dimer方法则通过构建一个包含两个相连原子的“二聚体”,并在势能面上进行旋转和移动来搜索过渡态。
三、量子化学过渡态模拟计算的应用
-
反应机理研究:通过计算过渡态的结构和能量,可以揭示化学反应的微观机理,包括键的断裂和生成、电子的转移和重排等过程。这对于理解反应的本质和预测反应条件对反应结果的影响具有重要意义。
-
催化剂设计:在催化反应中,过渡态的结构和能量往往决定了催化剂的活性和选择性。通过量子化学过渡态模拟计算,可以设计具有高效活性和选择性的催化剂,提高化学反应的效率和产率。
-
药物研发:在药物研发过程中,了解药物分子与生物靶标之间的相互作用机制至关重要。通过计算药物分子与生物靶标反应过程中的过渡态结构和能量,可以预测药物的活性和副作用,为药物设计和优化提供重要依据。
如您对所发信息感兴趣或是有需求的话,请联系信息发布人 联系我时,请说是在深圳华算科技有限公司看到的,谢谢!
