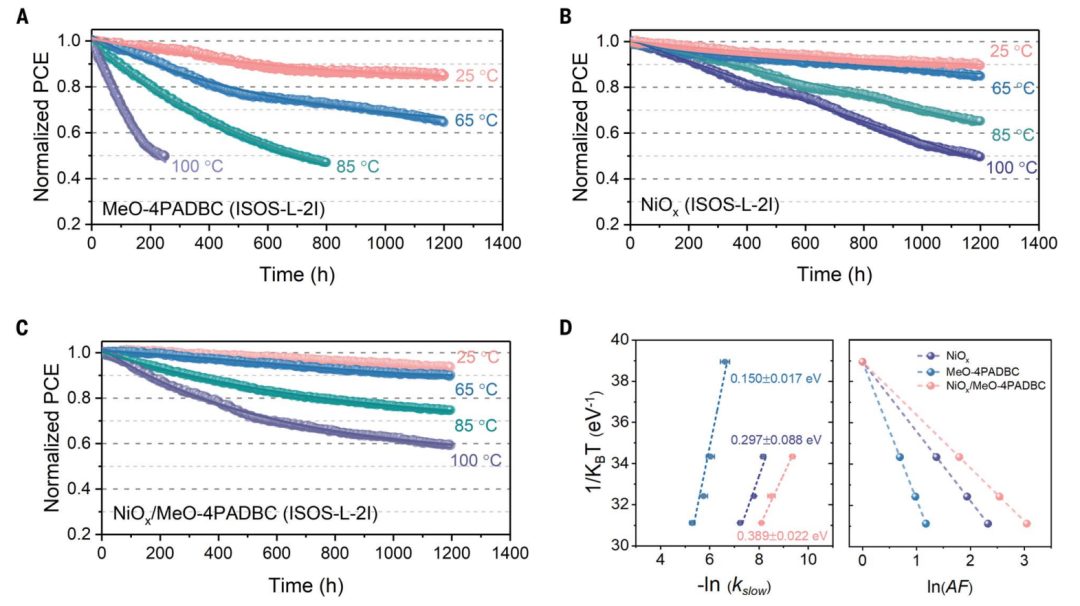
1.68和1.80 eV 宽带隙钙钛矿组合物器件也分别显示出令人鼓舞的22.7%和20.1%的PCE。此外,1.53 eV使用NiOx/MeO-4PADBC HSL的钙钛矿太阳能电池在65℃下能够循环1200小时,并保持90%的初始效率,和阿伦尼乌斯能量外推表明,太阳能电池在25°C能够保持80%的初始效率超过10个月。
相关研究成果以“Stabilized hole-selective layer for high-performance inverted p-i-n perovskite solar cells”为题发表在Science上。
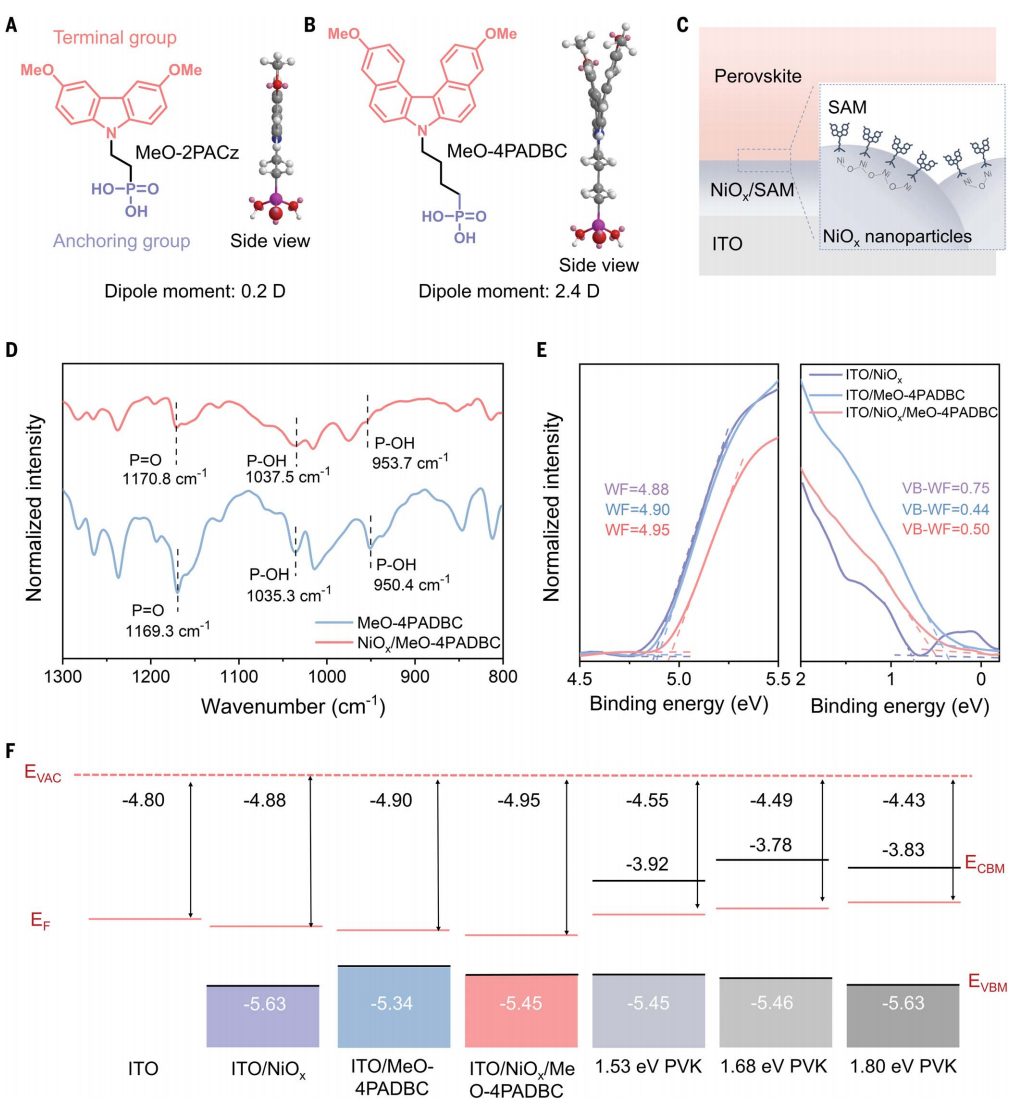
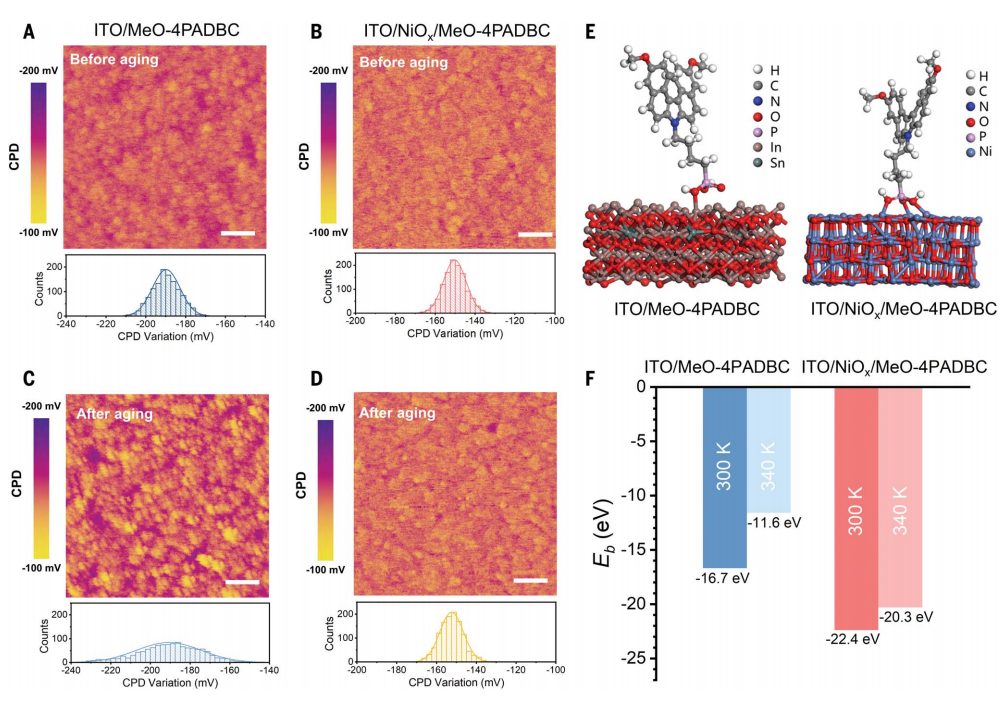
研究背景具有咔唑的膦酸自组装单层膜(SAMs)使钙钛矿太阳能电池(PSC)的性能得以提高,适用于单结和钙钛矿基串联太阳能电池,其具有高空穴选择性、快速空穴转移速率和低界面陷阱态密度。然而,与传统的聚合物和金属氧化物空穴传输材料相比,SAM基PSC表现出较差的热稳定性。在最大功率点(MPP)运行或开路电压(VOC)、高温(65°至85°C)下的运行稳定性,对于提高其稳定性和满足国际稳定标准至关重要。虽然大多数关于SAM基PSC的研究都报道了室温下的操作稳定性或通过稳定钙钛矿表面和体积增强了器件在热应力下的耐久性,但SAM形成分子在高温(>65°C)下的降解效应很少被讨论。SAM形成分子的热稳定性在很大程度上取决于它们与所选基底的结合,其锚定基团和分子间隔物之间的键可以通过温度诱导的解吸断开。内容详解SAM设计和合成调节末端官能团是调节SAM和钙钛矿之间界面相互作用的有效方法。例如,将两个甲氧基(OMe)引入2PACz中,改善了界面接触并使p-i-n PSC的效率更高。尽管如此,咔唑上的OMe取代也导致偶极矩从2PACz的~0.2D降低到MeO-0PACz的~2.2D,这反过来又导致SAM分子的最高占据分子轨道(HOMO)与钙钛矿的价带最大值(VBM)之间的偏移。这种现象可归因于咔唑结构的高平面度和对称性,掺入两个偶极矩方向相反的OMe基团使得MeO-2PACz的偶极矩接近于零(图1A)。作者通过使用非共面螺旋形二苯并[c,g]咔唑(DBC)单元作为核心来解决这个问题,以减少引入OMe基团时对偶极矩的负面影响,从而提供了MeO-4PADBC的新SAM(图1B)。同时,密度泛函理论(DFT)计算验证了MeO-4PADBC与(2-(4H-二苯并[c,g]咔唑-4-基)丁基)膦酸(7PADBC)(7.4D)相比,偶极矩(2.9D)略有降低,与咔唑基SAM相比具有较大差异。图1. HSLs的分子结构和电学性质空穴选择层应用作者比较了MeO-4PADBC SAM和NiOx/MeO-4PADBC作为p-i-n PSC的HSL(图1C)。然后通过傅里叶变换红外(FTIR)光谱表明化学键的形成。然而,沉积在ITO/NiOx/MeO-4PADBC衬底上的钙钛矿比沉积在对照衬底上具有更大的晶体域,这是因为SAM分子通过更强大的三齿酸结合吸收,将更密集的锚定在ITO/NiOx底物上,这不仅可以降低ITO/NiOx底物的表面粗糙度,但也导致了一个更疏水的表面和钙钛矿与SAM分子之间的相互作用更强。这些协同作用有助于促进钙钛矿晶体的成核和生长,从而促进钙钛矿的结晶。图2. 在不同的HSLs条件下,钙钛矿太阳能电池的光伏性能图3. 钙钛矿太阳能电池的降解机理分析图4. 不同温度下钙钛矿太阳能电池的长期稳定性评价综上所述,本文展示了一种高效且稳定的HSL,其热稳定性大大提高,适用于高效含SAM的倒置p-i-n PSC。MeO-4PADBC的合理分子结构设计和深入分析表明,最佳偶极矩和与钙钛矿的良好接触是理想能量排列和快速空穴提取提高器件效率和稳定性的关键。此外,将MeO-4PADBC SAM分子锚定在NiOx膜上,可以与NiOx形成更强的三齿键,有效降低电压损失,并在热应力下保持较强的固定效果。本文的研究为高效稳定的HSL设计提供了理论指导,并为轻松获得商业化倒置p-i-n PSC铺平了道路。Zhen Li†, Xianglang Sun†, Xiaopeng Zheng†, Bo Li†, Danpeng Gao, Shoufeng Zhang, Xin Wu,Shuai Li, Jianqiu Gong, Joseph M. Luther, Zhong’an Li*, Zonglong Zhu*, Stabilizedhole-selective layer for high-performance inverted p-i-n perovskite solar cells, Science, 2023,https://www.science.org/doi/10.1126/science.ade9637【做计算 找华算】华算科技专注DFT代算服务、正版商业软件版权、全职海归计算团队,10000+成功案例!Nature Catalysis、JACS、Angew.、AM、AEM、AFM等狂发顶刊,好评如潮!计算内容涉及材料结构、电位、容量、电导率、离子扩散,过渡态+AIMD、吸附、HOMO/LUMO、离子溶剂化配位结构、固态电解质锂离子通道、催化活性能、反应路径计算、OER、HER、ORR、自由能等。添加下方微信好友,立即咨询: