Jens Kehlet Nørskov,目前执教于丹麦技术大学物理系,曾是斯坦福大学SUNCAT界面科学与催化中心创始人(2010-2018)。Nørskov教授主要从事催化基础理论的相关研究,从电子层面出发,对催化反应规律进行描述,为催化剂设计做出了很多杰出贡献,近年来,在软件开发、算法研究、合成氨和燃料电池领域均有重大进展。特别是他提出的d-band center理论是目前计算化学领域应用最广泛的经典理论之一,推动了电催化领域的发展。截止目前,据Google Scholar统计,发表文章多达580余篇,总被引次数高达14万+,H-index为190。
J. Phys. Chem. Lett.: 乙腈-过渡金属界面的第一性原理研究
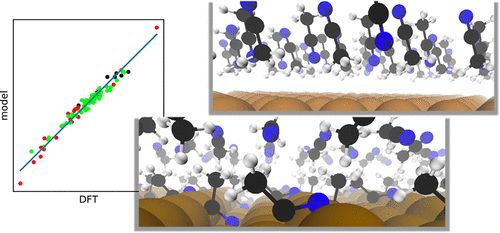
乙腈是催化和电化学中最常用的非水溶剂之一。本研究利用密度泛函理论研究了乙腈与金属Ag、Cu、Pt和Rh的多个晶面之间的界面,并强调了高覆盖率下溶剂-溶剂相互作用的重要性。研究了零电荷电位(potential of zero charge, PZC)与金属功函数的关系,发现了与实验测量一致的结果。并开发了一个模型来解释溶剂化学吸附和取向对PZC的影响,其平均绝对偏差为0.08–0.12 V。得到的静电场相图与光谱观察结果一致,为静电场效应的研究提供了新的思路。这项工作为乙腈-金属界面的实验观察提供了新的视角,并为乙腈和其他非水溶剂与过渡金属的界面研究提供了指导。
Acetonitrile Transition Metal Interfaces from First Principles, https://doi.org/10.1021/acs.jpclett.0c02692
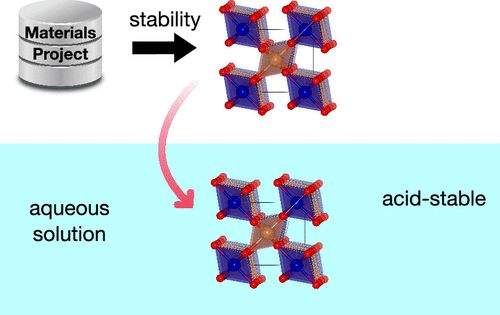
ACS Energy Lett.:ORR/OER条件下酸稳定氧化物周期表
评价材料在工作条件下的稳定性对器件导向型电催化剂的发展具有重要意义,但在这方面的研究却很少。在构建计算Pourbaix图的广泛工作的基础上,利用材料项目数据,探索了47 814种非二元金属氧化物在典型的ORR/OER条件下的水稳定性,并确定了68种可能的酸稳定的电催化剂,发现含有Sb/Ti/Sn/Ge/Mo/W元素的氧化物在酸性和氧化性环境中具有较高的耐蚀性。本研究还建立了一个“酸稳定周期表”,以指导寻找新的酸稳定电催化材料。
Acid-Stable Oxides for Oxygen Electrocatalysis,
https://doi.org/10.1021/acsenergylett.0c01625
ACS Catal.:钌基催化剂OER活性-稳定性关系
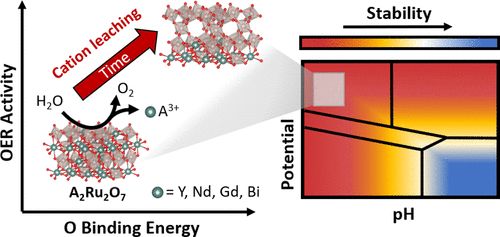
钌基析氧反应(OER)催化剂在高效电解水方面有着重要的应用前景。本研究对几种钌基焦绿石(A2Ru2O7,A=Y,Nd,Gd,Bi)作为OER催化剂进行了探索,并证明了相对于RuO2,Ru催化中心的活性和稳定性有所提高。结合补充的实验和理论分析,以了解A-位元素如何影响酸性OER条件下的活性和稳定性。在本文研究的A2Ru2O7中,结果发现Ru-O键较长,Ru4d和O2p轨道的相互作用弱于RuO2,导致初始活性增强。还观察到催化剂的OER活性随时间而发生变化,并伴随着A-位和Ru在不同相对速率下的溶解,这取决于A-位的特性。使用密度泛函理论(DFT)计算构建的Pourbaix图揭示了实验观察到的溶解驱动力,表明本文研究的所有成分在酸性OER条件下的热力学不稳定性。理论活性预测表明,A位阳离子浸出和OER活性之间的趋势一致。Bader电荷分析表明,溶解暴露出高度氧化的Ru位,显示出增强的活性。总的来说,使用稳定性指数(molO2 evolved/molRu dissolved)作为比较度量, A2Ru2O7材料显示出比标准RuO2更大的稳定性,并且与一些Ir混合金属氧化物具有相称的稳定性。本文所述的结论为提高钌催化剂的活性和耐久性,最终提高水电解槽的效率提供了途径。
Acidic Oxygen Evolution Reaction Activity–Stability Relationships in Ru-Based Pyrochlores, https://doi.org/10.1021/acscatal.0c02252
Chem Electro Chem:锂介导的合成氨反应研究
合成氨的电化学过程有可能取代Haber‐Bosch工艺。然而,N2是一种非活性分子,析氢反应是合成氨的主要选择性挑战。在四氢呋喃溶剂中电沉积锂的电极克服了这两个问题,它提供了一个容易与N2反应的表面,并限制了质子与非水电解质的接触。在这些条件下,本研究得到了相对较高的法拉第效率(约10 %)和速率(0.1 mA cm−2)。并观察到固体电解质界面层的发展以及锂和含锂物种的积累。DFT研究表明,由于氮化锂和氢化物在反应条件下的热力学和动力学稳定性以及N2在锂中的快速扩散,因此它们是催化活性相。
A Combined Theory‐Experiment Analysis of the Surface Species in Lithium‐Mediated NH3 Electrosynthesis, https://doi.org/10.1002/celc.201902124
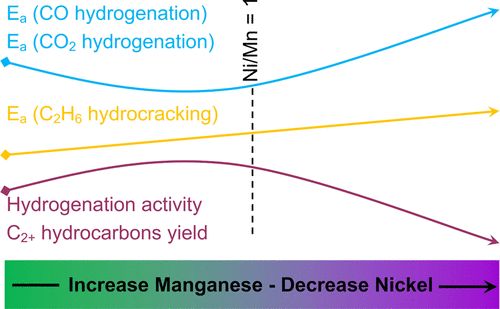
ACS Catal.:Mn对Ni/Al2O3催化剂CO/CO2加氢反应的促进作用
本文研究了Mn对Ni/Al2O3催化剂上CO和CO2加氢的促进作用。Ni/Al2O3对CO和CO2完全转化为甲烷和C2+烃具有较高的活性。此外,在离散且相对狭窄的温度范围内, C2+碳氢化合物的净浓度升高,出口流中含有比进料流中更高浓度的C2+物种,并且产品流几乎不含碳氧化物。结果表明,Mn的加入可以提高烃的选择性。本文研究了一系列不同Ni/Mn比的Ni-Mn/Al2O3催化剂,通过多种表征技术研究了Mn对烃产率的影响。通过密度泛函理论(DFT)的计算,揭示了Mn促进作用的来源,揭示了在还原条件下Ni(211)台阶边缘位置有利于Mn的置换。这些Mn物种对氧化物的亲和力稳定了共解离产物,因此提供了一个热力学驱动力,与未改性的Mn催化剂表面相比,促进了C–O键断裂。
Effect of Manganese on the Selective Catalytic Hydrogenation of COx in the Presence of Light Hydrocarbons Over Ni/Al2O3: An Experimental and Computational Study, https://doi.org/10.1021/acscatal.9b04863
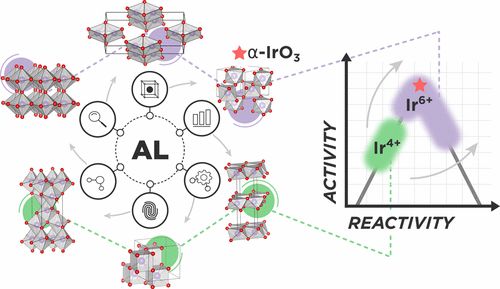
Chem. Mater.:主动学习算法用于发现OER稳定氧化铱多晶型
寻找高性能和稳定性的可持续能源应用材料是催化和材料科学的一个紧迫目标。本研究发展了一种易于推广的主动学习(active-learning, AL)加速算法,用于识别电化学稳定的IrO2和IrO3多晶型。该研究与随后的酸性OER结构的电化学稳定性分析相结合。通过在现有材料数据库(超过38000个)中识别所有956个结构上唯一的AB2和AB3原型来生成候选结构。使用主动学习的方法,在热力学非晶态合成极限内发现196个IrO2多晶型,并重新定义了金红石结构的全局稳定性。还发现了75种可合成的IrO3多晶型,并报道了一种先前未知的FeF3型结构,称为α-IrO3。为了测试算法的性能,对候选空间的随机搜索进行比较,发现率至少提高了2倍。另外,AL方法可以用少于30个密度泛函理论优化得到最稳定的IrO2和IrO3多晶型。对所发现的多晶型的结构性质的分析表明,几乎所有低能结构都首选八面体局部配位环境。随后的Pourbaix Ir–H2O分析表明,α-IrO3在酸性OER条件下是全局稳定的固相,并取代金红石IrO2的稳定性。本研究所提出的算法可以很容易地推广到任何二元金属氧化物结构的搜索并定义化学计量比。
Active Learning Accelerated Discovery of Stable Iridium Oxide Polymorphs for the Oxygen Evolution Reaction, https://doi.org/10.1021/acs.chemmater.0c01894
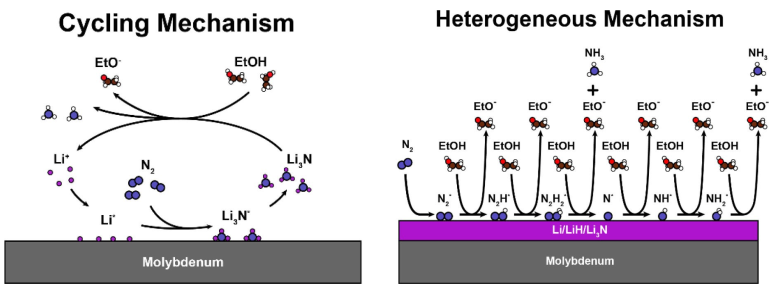
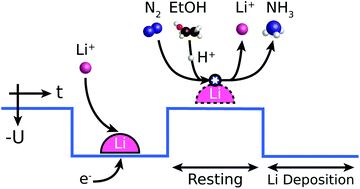
Energy Environ. Sci.:提高锂介导的电化学合成氨的稳定性和效率
锂介导的氮还原是一种已被证实的电化学合成氨的方法。本研究提出了一个通用的分子水平模型来描述锂离子介导的N2还原成NH3的机理。这个模型可以解释一些已经被实验观察到的机械特性,例如法拉第效率对N2分压和EtOH浓度的依赖性。基于这一认识,开发了一种新的增加法拉第效率的方法,即在有利于锂沉积和有利于锂溶解的范围之间循环施加电位。该方法可显著提高体系的稳定性和催化效率。实验表明,锂循环过程在长达(至少)125小时的运行中是稳定的,并且在潜在循环期间,法拉第效率从恒定的锂沉积期间的∼21%大幅增加到∼37%。测量到了∼7%的能量效率,远远超过了文献中短期6分钟实验中2.8%的最高值。
Increasing stability, efficiency, and fundamental understanding of lithium-mediated electrochemical nitrogen reduction, https://doi.org/10.1039/D0EE02246B
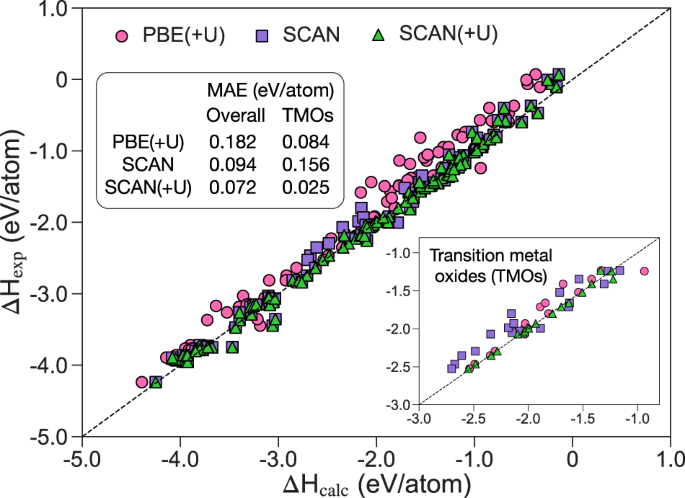
npj Comput Mater:用SCAN泛函计算Pourbaix图来预测固体的水稳定性
本研究中利用扫描函数,在材料工程(MP)Pourbaix图框架上开发了一种简单的方法来精确预测固体的水稳定性。评估了扫描函数在计算各种氧化物生成焓方面的性能,并对标准扫描函数显示出较大偏差的过渡金属氧化物进行了Hubbard U校正。通过与实验和MP-PBE-Pourbaix图的比较,验证了用扫描函数计算的Pourbaix图的性能。基准测试表明,SCAN-Pourbaix图在水稳定性预测方面系统地优于MP-PBE。进一步说明了该方法在准确预测酸性介质中析氧反应催化剂溶解势方面的应用。
Predicting aqueous stability of solid with computed Pourbaix diagram using SCAN functional, https://doi.org/10.1038/s41524-020-00430-3
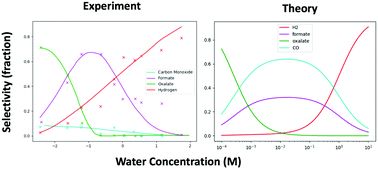
Phys. Chem. Chem. Phys.:非水溶剂中Pt电极上CO2RR的微观动力学模型
析氢(HER)反应与二氧化碳电化学还原(CO2RR)制备多碳产品之间的竞争是众所周知的挑战。本研究提出了一个简单的微观动力学模型,在强还原电位下,这些竞争反应在Pt催化剂上,在非水溶剂中具有不同的质子浓度。该模型提供了对反应机理的一些见解,并表明低质子浓度和高阶跃位置可能会提高对多碳产物的选择性。
Micro-kinetic model of electrochemical carbon dioxide reduction over platinum in non-aqueous solvents, https://doi.org/10.1039/C9CP05751J
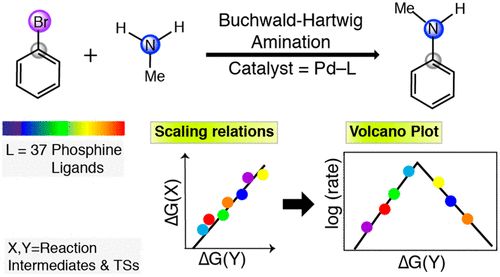
ACS Catal.:均相催化Buchwald–Hartwig amination中的标度关系分析
标度关系(Scaling relations)广泛应用于研究表面反应。然而,对其在均相催化中的应用的探索目前还鲜有报道。本文研究了标度关系概念对Buchwald–Hartwig amination (BHA)反应的均相催化反应的可转移性。用37种不同的Pd–L配合物(L = phosphine ligand)研究了PhBr与MeNH2的反应,建立了反应中间体与过渡态之间的标度关系。用能量图的中间比例来构造火山的比例关系。火山图的结果与文献中的实验趋势很好地吻合,这为更好地设计催化剂提供了方向。
Scaling Relations in Homogeneous Catalysis: Analyzing the Buchwald–Hartwig Amination Reaction, https://doi.org/10.1021/acscatal.9b04323
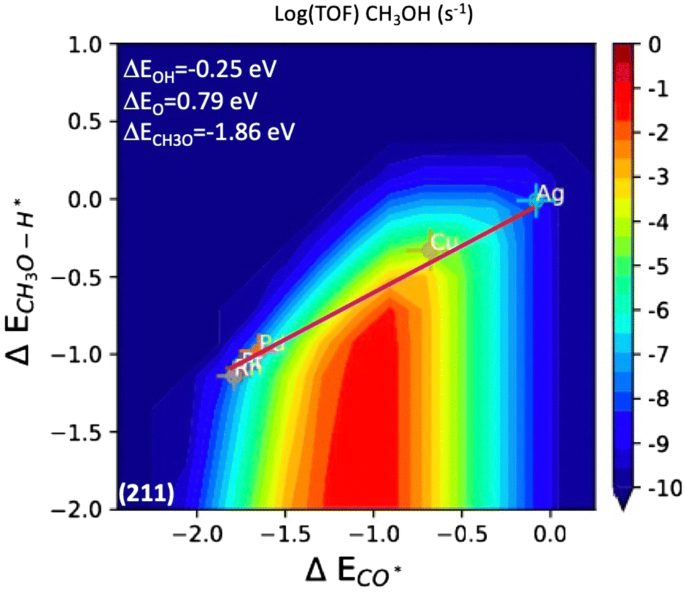
Top. Catal.: CO加氢制甲醇的挑战揭示线性标度关系的基本限制
近年来计算催化技术的发展使得基于线性标度关系的复杂反应网络维数的常规降维方法得以实现。本研究对CO加氢制甲醇活性的基本限制进行了新的描述;这一反应为从合成气中获得增值化学品提供了一条可持续的途径。首先,证明了CO*的生成能(其中*表示吸附物种)与这些表面上甲醇合成路径上一些基本步骤的过渡态能量之间有很强的线性相关性。利用微观动力学模型,将这些信息投射到活动火山图中,以给定过渡态的形成能和CO*作为独立的描述符。这一分析揭示了上述线性标度关系对活性的基本限制,并促使人们积极寻找新的材料,以摆脱这些线性标度关系,作为提高CO加氢制甲醇活性的必要条件。本文还指出H–CO*和CH3O–H*是独立于CO*稳定的关键过渡态,可以提高甲醇合成的活性和选择性。
The Challenge of CO Hydrogenation to Methanol: Fundamental Limitations Imposed by Linear Scaling Relations, https://doi.org/10.1007/s11244-020-01283-2
原创文章,作者:Gloria,如若转载,请注明来源华算科技,注明出处:https://www.v-suan.com/index.php/2023/10/24/1a96547f31/