由锂盐、聚合物基体和溶剂组成的准固态聚合物电解质(QPE)有利于提高电池的安全性和能量密度。然而,QPE的离子传导机制、溶剂分子的存在形式以及不同组分之间的相互作用仍不清楚。
近日,南开大学陈军院士等人开发了一种多光谱表征策略,结合第一性原理计算通过分析具有优异电化学稳定性的聚偏二氟乙烯六氟丙烯(PVDF-HFP)模型体系来解开上述谜团。结果表明,QPE中溶剂的存在状态与液态电解液中的溶剂存在状态有很大不同。部分溶剂分子在QPE中使聚合物基体和锂盐之间形成空间隔离,而另一部分溶剂用于溶解锂盐以形成局部高浓度的Li+。因此,Li+的溶剂化结构和导电机制与高浓度液态电解液中的相似。
这项工作为QPE的离子传导机制提供了新的见解,并将促进其在安全和高能电池中的应用。相关成果以题为“A new insight into the ionic conduction mechanism of quasi-solid polymer electrolyte through multispectral characterizations”发表在Angew. Chem. Int. Ed.上。
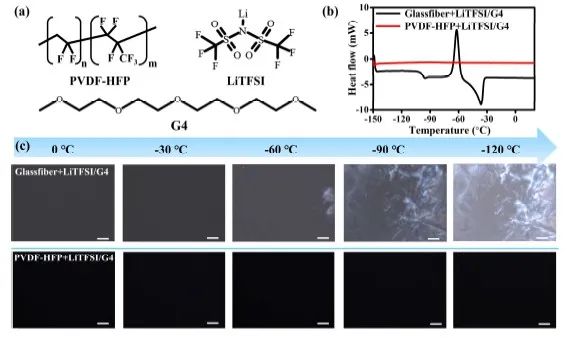
这项工作的研究体系由PVDF-HFP、LiTFSI和四甘醇(G4)组成。为探究QPE和液态电解液之间G4的存在状态差异,首先通过DSC对其相变进行了表征。研究显示,G4在玻纤+LiTFSI/G4中的凝固点约为-46°C(图1b),接近于纯G4液体(-30°C)。然而,QPE在-150 °C 到25 °C范围内没有相变,这表明QPE中存在G4的过冷现象,这意味着PVDF-HFP与G4之间存在强相互作用。同时,采用偏光显微镜(图1c)研究冷冻前后G4溶剂的各向同性和各向异性。随着温度从0 °C下降到-120 °C,玻纤+LiTFSI/G4中的G4呈现出明显的从非晶相液相到晶相固体的相变,与之前的DSC数据一致。然而,QPE中的G4没有表现出任何相变并随着温度的变化保持非晶相。基于以上分析,可以得出结论,QPE中G4的存在状态与液态LiTFSI/G4中的存在状态有很大不同。
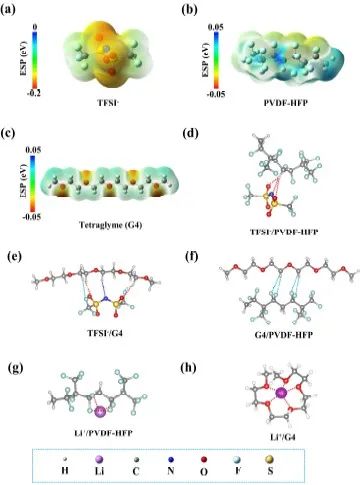
为探索QPE体系的相互作用并降低结构和组成的复杂性,作者首先计算了各组分的静电势(ESP)(图2a-c)和双组分体系中相互作用的优化几何构型(图2d-h)。双组分体系中官能团的键长变化是各组分间存在相互作用的证据。不同原子的ESP表现出很大的多样性,这有助于负电和正电原子之间的配位,导致相应官能团的键长发生变化。同时,键长的变化会对特征峰波数产生影响。
图2 各组分的ESP和双组分体系中相互作用的优化几何构型
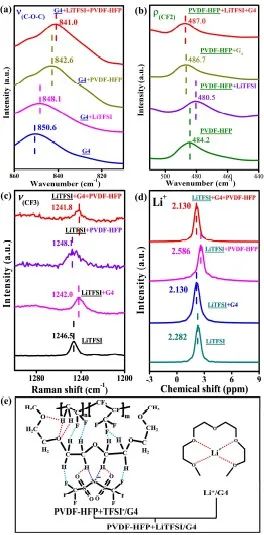
首先作者通过FTIR选择G4中C-O-C官能团的伸缩振动(ν(C-O-C))来研究G4与LiTFSI或PVDF-HFP的相互作用。与纯G4(图3a)相比,G4+LiTFSI和G4+PVDF-HFP体系中的ν(C-O-C)偏差值约分别为−2.5 cm-1和−8.0 cm-1。官能团键长的增加导致其波数的减少,反之亦然。这意味着在LiTFSI/G4和PVDF-HFP/G4体系中,G4中C-O-C的键长呈现出增加的趋势。这一结论与上述计算的几何构型相一致。对于LiTFSI/G4,G4的O原子和Li+之间的配位相互作用(图2h),以及TFSI-的F、N、O原子和G4的H原子之间的的静电相互作用(图2e),使C-O-C的键长分别从1.41410Å增加到1.43061Å和1.41738Å。对于G4/PVDF-HFP,C-O-C的键长增加是由于F原子(PVDF-HFP中)和H原子(G4中)之间的静电相互作用,如图2f所示,这使得C-O-C的键长从1.41410Å增加到1.41671Å。值得注意的是,QPE中ν(C-O-C)位置的偏差值约为−9.6 cm-1,大于G4+LiTFSI或G4+PVDF-HFP。因此可以推断,QPE中的G4同时接收LiTFSI和PVDF-HFP的静电相互作用。
进一步选择PVDF-HFP中-CF2的摇摆振动 (ρ(-CF2)) 来研究PVDF-HFP与LiTFSI或G4的相互作用。与纯PVDF-HFP相比,PVDF-HFP+LiTFSI和PVDF-HFP+G4体系中ρ(-CF2)的偏差值分别约为-3.7 cm-1和2.5 cm-1(图 3b)。值得注意的是,QPE中ρ(-CF2)位置的偏差值约为2.8 cm-1,与 PVDF-HFP+ G4非常接近。因此,很容易推测QPE中的PVDF-HFP组分仅受G4组分的影响,这意味着LiTFSI和QPE中的PVDF-HFP组分之间没有明显的相互作用。
进一步,采用拉曼光谱以研究LiTFSI与G4或PVDF-HFP的相互作用。与LiTFSI 相比,LiTFSI+G4和LiTFSI+PVDF-HFP 中ν(-CF3)的偏差值分别约为-4.5 cm-1和1.6 cm-1(图 3c)。LiTFSI+G4中G4的引入使得ν(-CF3)的吸收峰向较低波数移动,这意味着相应官能团的键长增加。这可能是由于TFSI-/G4体系TFSI-的N、F、O原子和G4的H 原子之间的静电相互作用。然而,在LiTFSI+PVDF-HFP中引入PVDF-HFP使得ν(-CF3)吸收峰向更高波数移动。值得注意的是,QPE中 ν(-CF3)位置的偏差值约为-4.7 cm-1,非常接近LiTFSI/G4。因此,很容易推测TFSI-组分只受G4的影响,即TFSI-和PVDF-HFP组分在QPE中没有明显的相互作用。
核磁共振(NMR)可以反映Li+的配位环境并分析相关的溶剂化结构。研究显示,与LiTFSI相比,LiTFSI+G4和LiTFSI+PVDF-HFP体系中Li峰的偏差值分别约为-0.152 ppm和0.304 ppm。这两个体系显示出相反的偏差方向,这意味着G4和PVDF-HFP对Li+的影响截然不同。有趣的是,QPE中Li峰的偏差值也是-0.152 ppm,与LiTFSI/G4相同。也就是说,Li+的溶剂化结构在这两个体系中也是相似的。根据上述拉曼和核磁共振结果,可以推测Li+和TFSI-仅受QPE中G4成分的影响。因此,在QPE中,PVDF-HFP与Li+或TFSI-没有明显的相互作用。
基于上述结果,可以肯定地推断出PVDF-HFP在QPE中与部分G4溶剂分子完全结合,导致游离G4溶剂分子数量减少,抑制凝固,提高Li+浓度。此外,NMR结果表明LiTFSI/G4和QPE中Li+的溶剂化结构和配位数相似。这意味着QPE 中G4分子的另一部分是自由分子,用于溶解锂盐,就像在液态电解液中一样,形成局部高浓度的Li+以确保快速离子传输。
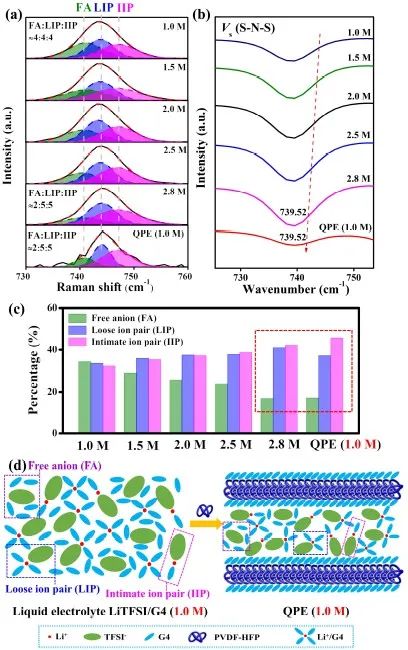
为进一步验证这一推论并分析溶剂化结构的差异,作者通过LiTFSI/G4和QPE 体系中的拉曼光谱探索了阴阳离子的相互作用。首先测试了不同浓度的LiTFSI/G4溶液,范围从 1.0 M 到 2.8 M。随着LiTFSI浓度的增加,游离阴离子(FA)比例减少,而松散离子对(LIP)和紧密离子对(IIP)比例均增加。而QPE的阴阳离子配位结构与浓度为2.8 M的LiTFSI/G4非常相似。这进一步验证了QPE的Li+的溶剂化结构和导电机制与高浓度液态电解液的相似。这一结论也得到了FTIR光谱数据的验证,相应的表征如图4b所示。
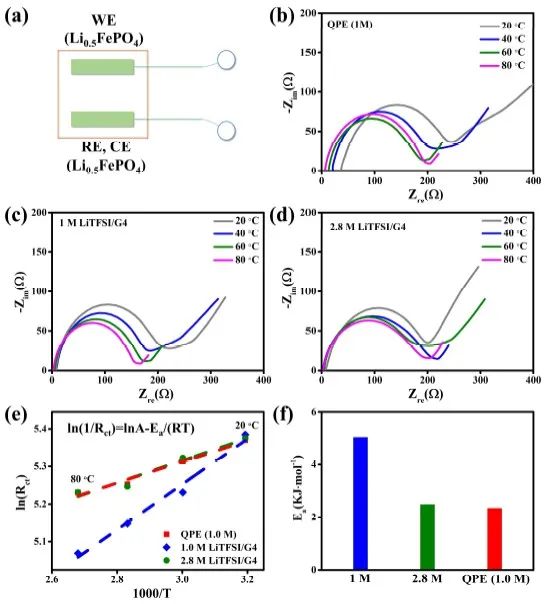
为进一步验证QPE中Li+溶剂化模式的正确性,作者评估了QPE中Li+溶剂化/去溶剂化过程的活化能 (Ea),并与1.0 M和2.8 M LiTFSI/G4液态电解液进行对比。结果,QPE、1.0 M 和 2.8 M LiTFSI/G4液态电解液中溶剂化/去溶剂化过程的Ea分别为2.345、5.038和2.485 kJ·mol-1(图 5f)。QPE中的Ea值与浓度为 2.8 M 的LiTFSI/G4非常接近,这证实了QPE的Li+溶剂化模式。
上述结论对准固态电解质的设计有一定的启发作用。例如,为提高QPE的热稳定性,可以根据相似性互溶理论,用一些类似的溶剂官能团修饰聚合物基体,以增强聚合物基体与溶剂之间的相互作用。这种相互作用策略还可以用来改变捕获的强相关溶剂分子和自由分子的比率,以调节Li+浓度和离子电导率。
总之,作者使用多光谱表征策略探索了一种基于PVDF-HFP的经典QPE的离子传导原理和电解质结构。研究表明,QPE中溶剂的存在状态与传统液态电解液中的存在状态有很大不同。QPE中的部分溶剂分子通过强相互作用与PVDF-HFP 完全结合,而其他溶剂分子则用于溶解锂盐并形成局部高浓度Li+ 以确保快速离子传输。
A new insight into the ionic conduction mechanism of quasi-solid polymer electrolyte through multispectral characterizations. Angew. Chem. Int. Ed. 2021. DOI: 10.1002/anie.202107648
原创文章,作者:科研小搬砖,如若转载,请注明来源华算科技,注明出处:https://www.v-suan.com/index.php/2023/10/25/121d5b4181/