

II–VI族和III–V族的胶体量子点(QDs)是下一代发光器件的关键成分。传统的II – VI族和III – V族半导体化合物(CIIAVI和CIIIAV,其中C代表阳离子,A代表阴离子)可以结晶成稳定的立方锌-闪锌矿结构。由于具有四面体键的特性,它们也被称为四面体半导体。利用了锌-闪锌矿结构的松散堆积特性,部分或全部占据,可以形成“填充四面体化合物”这些四面体半导体的功能衍生物的普遍设计原则是所谓的“填充四面体”,这些衍生材料由于其独特的特性,包括光电子学、压电学、热电学和自旋电子学,在广泛的应用领域展示了它们的巨大潜力。
河南大学曾在平团队为四面体半导体及其功能衍生物提供了另一种设计原则,即阳离子稳定带电簇网络。在这一原理的指导下,预测了三类新的立方材料,即多孔二元化合物、I–II–VI三元化合物和I–II-III–V四元化合物。利用第一性原理计算,从理论上筛选出65种高度稳定的候选材料。它们的结构和成分多样性使得发射波长从远红外到紫外区域,具有广泛的可调性。这项工作丰富了四面体半导体及其衍生物家族,可能对广泛的光电子应用领域感兴趣。
所有的计算都是在密度泛函理论(DFT)的框架内使用第一性原理方法进行的,正如维也纳从头算模拟包(VASP)所实现的那样。几何优化是通过使用Perdew-Burke-Ernzerhof (PBE)类型的广义梯度近似(GGA)以及投影缀加平面波(PAW)赝势来完成的。
在几何优化过程中,模拟槽内原子的位置、晶格参数以及相应的角度被完全放松,直到原子力达到0.01 eV/Å的公差。选择动能截止值为500 eV,能量收敛准则为10−5 eV/cell。布里渊区使用6 × 6 × 6 Γ-centered Monkhorst-Pack网格进行采样。声子谱的计算采用密度泛函微扰理论(DFPT),采用声子包,输入力由VASP代码计算。
本文利用Nose-Hoover恒温器对典型系综(NVT)内的2 × 2 × 2超级单体进行从头算分子动力学(AIMD)模拟。时间步长为2 fs,模拟总时间为20 ps,使用Γ-point对所有模型的布里渊区进行采样。VASP采用正常的模拟设置。对波函数的部分占比采用高斯涂抹,涂抹参数为0.05 eV。
电子最小化的收敛准则为10−4 eV/cell。利用HSE06函数对电子能带结构进行了计算,给出了所探索功能材料带隙的合理估计。HSE06函数中精确Hartree-Fock交换的部分选择为默认值,α = 0.25,筛选参数ω = 0.11玻尔−1。HSE06计算中的k-mesh选择4 × 4 × 4 Γ-centered Monkhorst-Pack mesh,其收敛带隙的精度在0.1 eV以下。
本文考虑了三种情况:(i)[CIIA4VI]6−簇的对抗是二价碱土(IIA组)或过渡金属(IIB组),而[CIIIA4V]9−簇的对抗是IIIA组的三价元素。
所有这些阳离子都只占据六个面中心的晶格点(图 1b);(ii)带电团簇[CA4]是II−VI组(即,[CIIA4VI]6−),而对位物是一价碱金属,同时占据6个面中心和12个边缘中心的晶格点(图 1c);(iii)带电簇[CA4]为III−V组(即[CIIIA4V]9−),而对抗是一价碱金属(IA组)和二价碱土金属(IIA组)或过渡金属(IIB组)的混合组合。一价阳离子占据6个面心晶格点,二价阳离子占据12个边心晶格点,反之亦然(图 1d)。
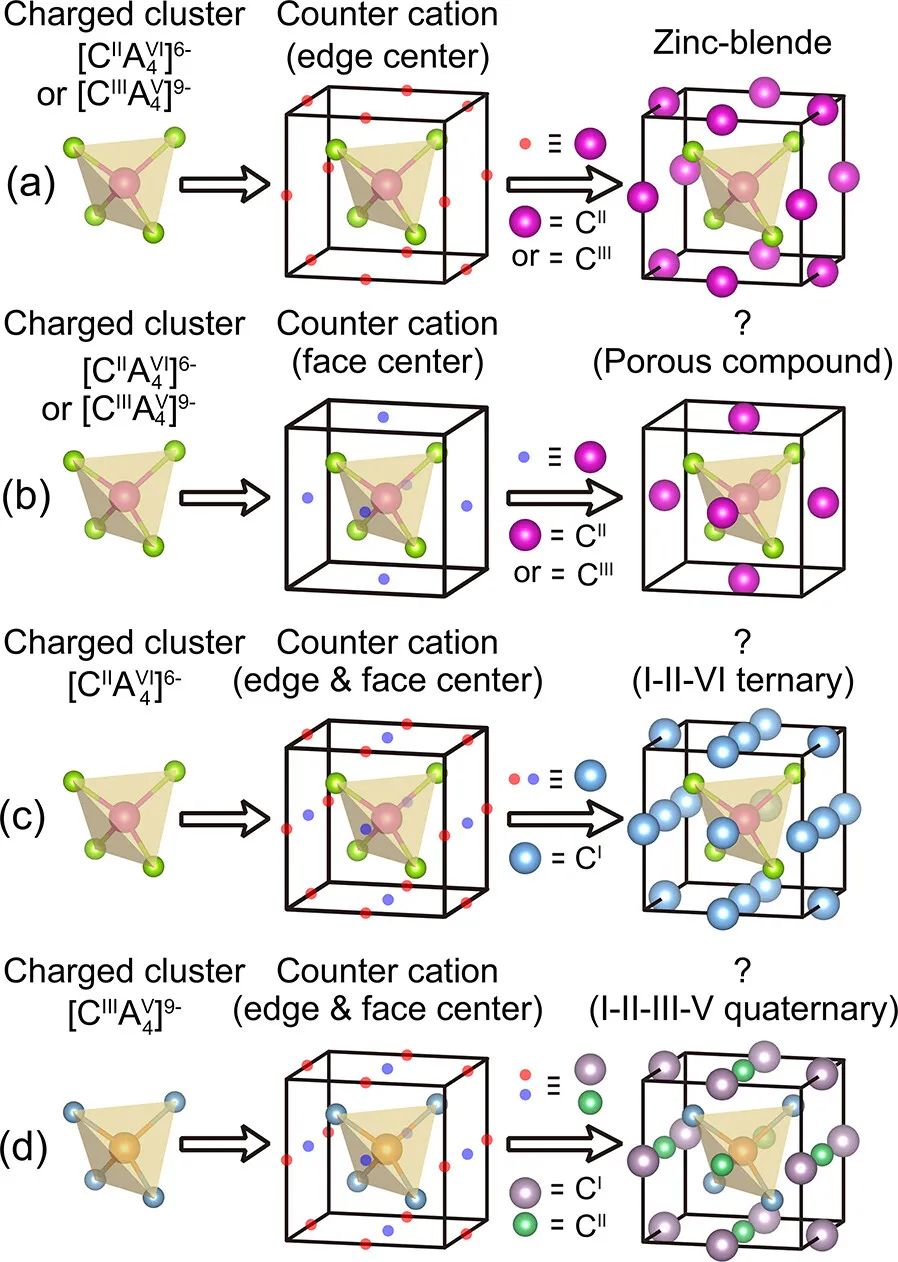
图1. 基于阳离子稳定带电簇网络原理的四面体半导体及其功能衍生物的设计方案,(a)锌闪锌矿结构在立方晶胞体中心放置一个带电团簇[CIIA4VI] 6−(或[CIIA4V] 9−),并在12个边中心晶格点处放置[CII](或[CIII])离子;(b)与(a)相似,但将反阳离子移动到六个面心晶格点导致多孔化合物的形成;(c)将[CIIA4VI] 6−簇置于体中心,在边中心和面中心晶格点放置单价阳离子CI;(d)与(c)相似,由[CIIA4V] 9−聚集在体中心,而单价(CI)和二价(CII)元素分别占据边中心和面中心晶格点
为了探究闪锌矿结构电子性质,II组−VI中的多孔化合物被发现是半导体或绝缘体,其发射波长从可见区域到紫外区域(图2a)。这些化合物的显著特征之一是,它们几乎对所有高度稳定的II−VI类和III−V类成员表现出直接带隙性质(图2a、b)。
闪锌矿结构不是这样,其中硫系化铍(BeS,BeSe,BeTe;图2c),AlAs和AlP是众所周知的间接半导体。与II组−VI组相比,它们在多孔结构和闪锌矿结构之间的带隙上表现出更大的能量差异。以阴离子为中心的多孔结构比以阳离子为中心的多孔结构具有更小的带隙,我们排除了沿着这条路线设计的设计材料。
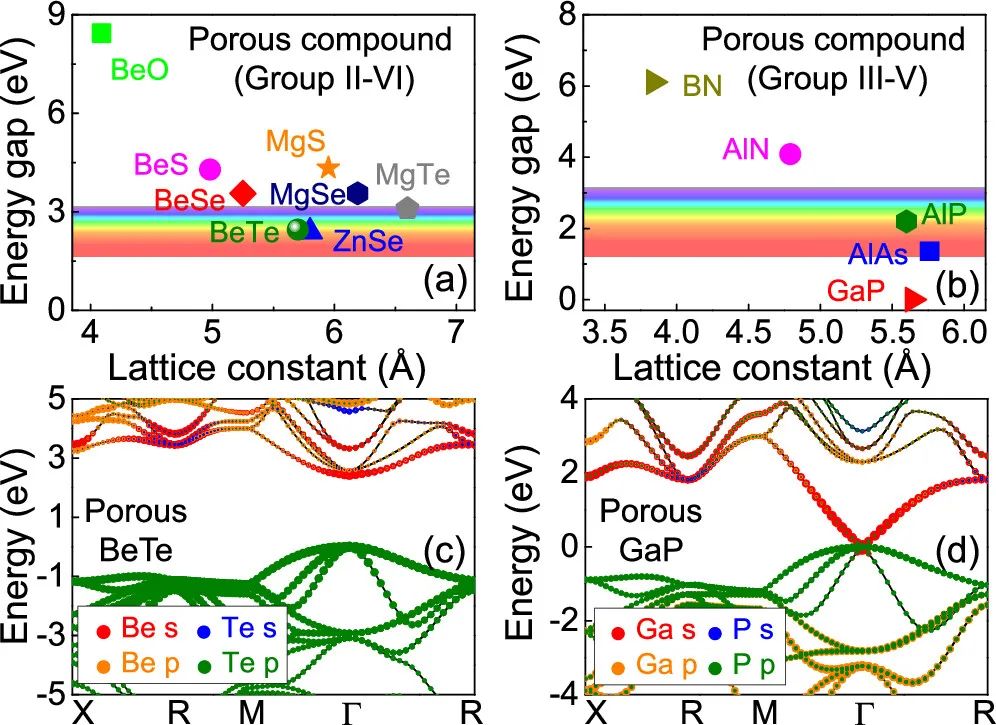
图2 (a) 8个第II ~ VI族稳定多孔化合物和;(b) 5个第III ~ V族稳定多孔化合物的带隙与计算晶格常数的关系;(c) II – VI族的多孔BeTe;(d) III – V族的GaP的能带结构
按照设计多孔化合物的相同配方,我们考虑了由CI =(Li、Na、K、Rb、Cs)、CII =(Be、Mg、Ca、Sr、Ba、Zn、Cd)和AVI =(O、S、Se、Te)组成的成员;因此总共有140种候选材料。
在热力学上发现,对于给定的带电团簇网络,较轻的抵消有利于更好的热力学稳定性。通过测试发现三元化合物变得不稳定的临界温度为对于LiZnS,Tc≈1000K(图 3a)和对于RbZnS,Tc<100K。通过设置Tc > 800 K,我们筛选了11个高度稳定的三元化合物(图3a和图4),如图3b所示。
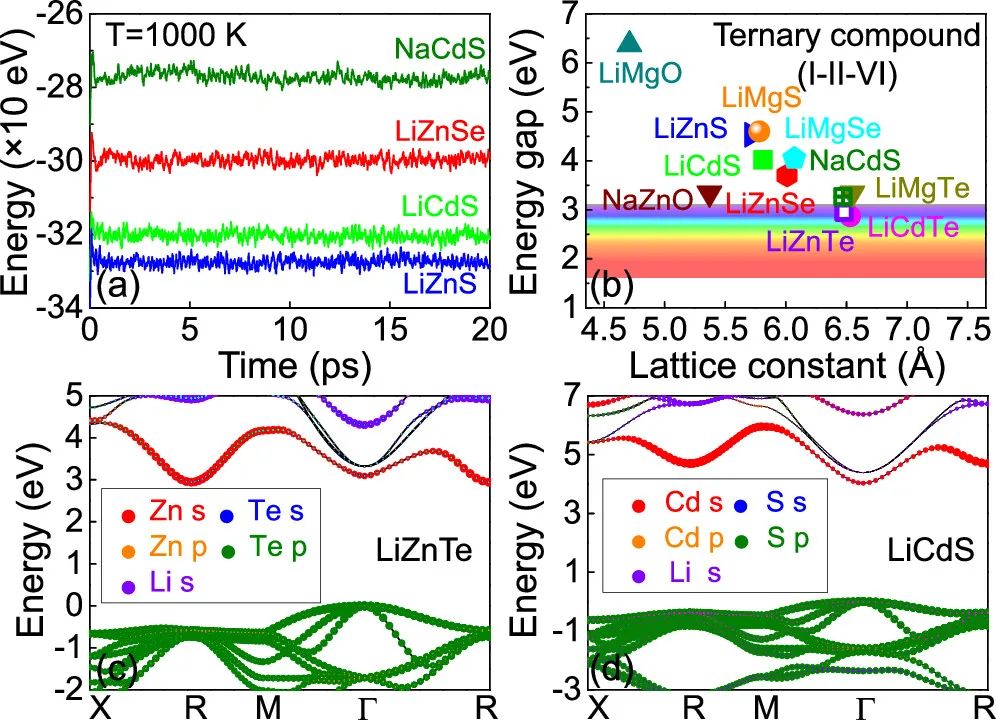
图3. (a)从头算分子动力学模拟(AIMD)的四个例子C6ICIIA4VI三元化合物,即NaCdS,LiZnSeLiCdS,和LiZnS,在温度T=1000k;(b)11个D在FT/HSE06水平的理论计算的稳定C6ICIIA4VI三元化合物的带隙与晶格常数的关系;(c) LiZnTe和(d) LiCdS分别在DFT/ HSE06理论计算的能带结构
图4. I-II-VI三元化合物的3×3×3超级单体,以及7个选定的候选物在高温20 ps下的从头算分子动力学(AIMD)模拟
当I族的阳离子变重时,三元化合物的晶格常数增加,能隙减小(如图3b中的LiCdS和NaCdS),与预期一致。一般来说,几乎所有被探索的热力学高度稳定的三元化合物都是在蓝光或紫外区域发射波长的直接带隙半导体或绝缘体(图3b)。
通过同时满足振动的标准和机械稳定性,以及在温度超过800 K时的热力学稳定性,作者筛选了41种高度稳定的材料。其中,21种属于前一类(CICIICIIIAV),以及那些具有直接间隙性质(共13种材料)选择性地显示在图5(a)中。其余的20个成员都属于后一类(CICIICIIIAV;),以及直接间隙材料(共10种材料)被显示在图5b中。

图5.(a,b)23个最佳的稳定结构的能量带隙,(a) CICIICIIIAV(总共13)和(b) CICIICIIIAV(总共10)化合物直接带隙性质和化学计量3−3−1−4相应的晶格常数的函数,强调表明独特的阳离子元素占据的晶格点;(c,d)分别在DFT/HSE06理论水平上计算的(c) LiZnAlAs和(d) LiZnAlP的能带结构示例
综上所述,本文介绍了一种设计四面体半导体及其函数导物的新原理,即阳离子稳定带电簇网络。它的预测能力已经被发现了三种新的立方材料,即多孔二元化合物、I−II−VI三元化合物和I−II−III−V四级化合物。
在这一原理的指导下,在332种候选材料中有65种高稳定材料从理论上进行了初步计算,显示出从远红外到紫外区域的可调发射波长。
未来的工作可能会扩展到探索它们在纳米尺度上的物理、电子和光学特性,以及在热电学和自旋电子学领域的潜在应用。这项工作极大地丰富了四面体半导体及其衍生物的家族,这可能在未来的实验实现中具有广泛的实际应用价值。
Min J, Zhai J, Dong T, et al. Design Principle for Tetrahedral Semiconductors and Their Functional Derivatives: Cation Stabilizing Charged Cluster Network[J]. Nano Letters, 2023.
https://pubs.acs.org/doi/10.1021/acs.nanolett.3c01352
原创文章,作者:v-suan,如若转载,请注明来源华算科技,注明出处:https://www.v-suan.com/index.php/2023/10/25/884be1f17e/

