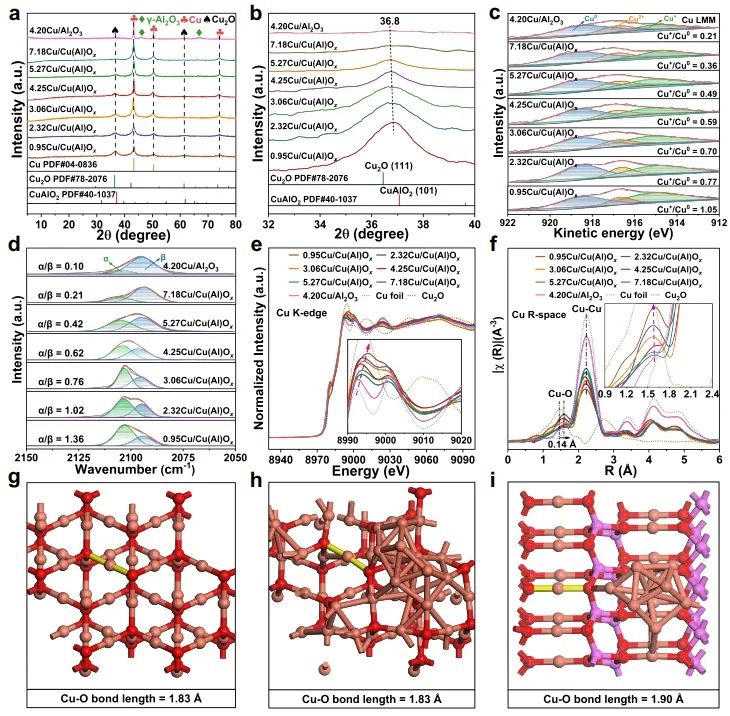
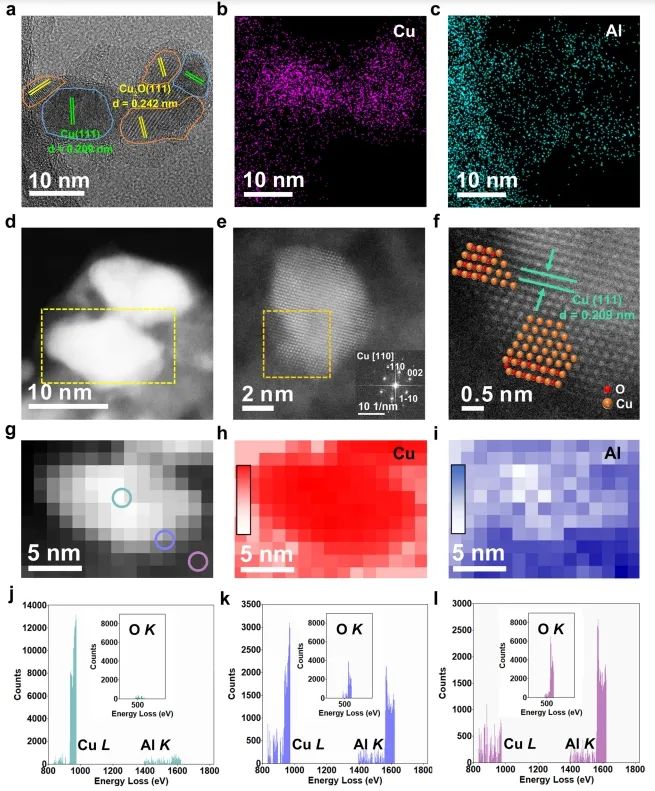
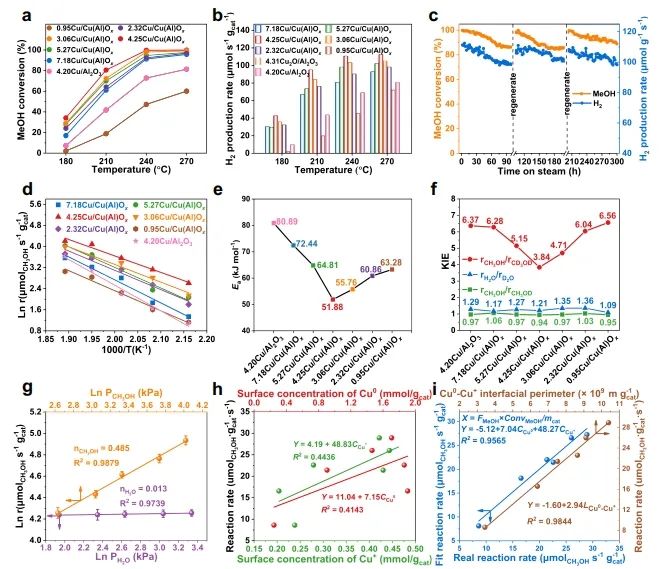
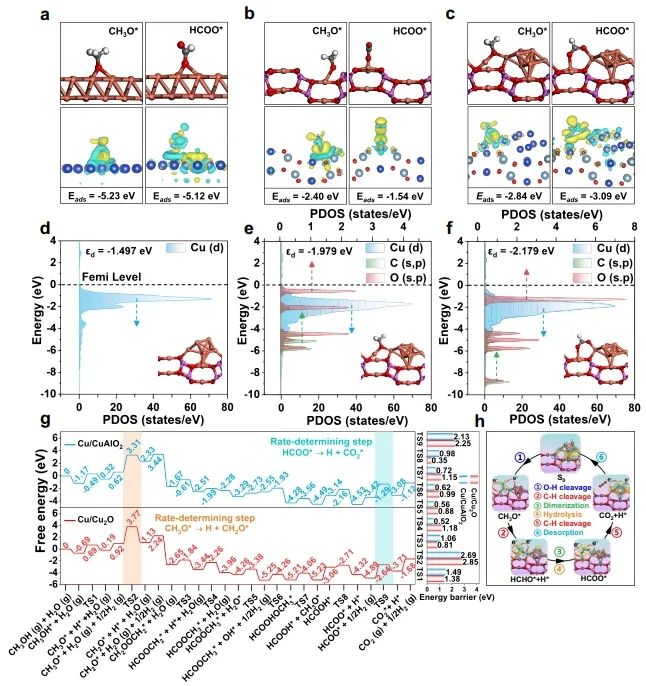
1. Nat. Commun.: 构建Cu0-Cu+双位点,有效改善MSR反应中C-H键断裂 随着资源和环境问题的日益严峻,氢能被认为是化石能源的有效替代品。其中,甲醇(CH3OH)水蒸气重整(MSR)由于能耗较低和操作简单而成为一种经济有效的产氢方法。在催化MSR反应的各种催化剂中,Cu具有成本效益、良好的低温活性和高H2选择性等优点。由于Cu具有丰富的氧化还原性质,在实际反应条件下,多种铜物种(Cu0,Cuδ+/Cu+)通常共存;此外,合金中的电子重排效应、强烈的金属-载体相互作用(SMSI)和氧空位诱导使得揭示本征活性位点、吸附行为和反应机理等基础问题相当复杂,导致催化性能与微观结构之间的关系不明确。因此,揭示催化剂的本征活性位点、构效关系和反应机理不仅可以为多相催化剂的结构设计提供合理的依据,而且可以促进MSR实际应用的进一步发展。近日,北京化工大学卫敏、张建、杨宇森和浙江大学肖丰收等通过共沉淀法和后续还原处理制备了一系列具有可调Cu0-Cu+位点的yCu/Cu(Al)Ox样品(y表示Cu/Al的质量比),并通过原位表征和理论计算研究了催化剂的活性中心。实验结果表明,在240 ℃下,4.25Cu/Cu(Al)Ox催化剂表现出最佳的MSR催化性能,CH3OH转化率>99%,H2产率高达110.8 μmol s-1 gcat-1,这优于先前报道的用于MSR的Cu基催化剂。此外,4.25Cu/Cu(Al)Ox催化剂的CH3OH转化率和H2产率在100 h后有所下降(分别从99.5%和110.8 μmol s-1 gcat-1降至86.3%和99.4 μmol s-1 gcat-1),但经过再生(300 ℃空气氧化1 h,220 ℃下25% H2/N2还原1 h)后,催化性能可恢复到原来的水平。动力学研究、原位FT-IR光谱和质谱法分析证实,4.25Cu/Cu(Al)Ox催化剂上的MSR反应经历三个主要过程:CH3OH脱氢、HCOOCH3水解和HCOO*分解,其中CH3O*和HCOO*中间体的C-H键断裂是速率控制步骤。值得注意的是,Cu0-Cu+界面协同催化起决定性作用:含氧中间体(CH3O*和HCOO*)在Cu0-Cu+界面位置发生吸附活化,适中的吸附强度引起催化剂界面的重构以及从催化剂界面到反应中间体的电子转移;几何结构和电子结构的变化导致C-H键断裂的能垒降低,显著促进了MSR反应。综上,该项工作在原子水平上揭示了MSR中Cu0-Cu+界面的协同效应,这可能为合理设计高性能的MSR催化剂提供指导。Designing Cu0−Cu+ dual sites for improved C−H bond fracture towards methanol steam reforming. Nature Communications, 2023. DOI: 10.1038/s41467-023-43679-0 2. Nat. Commun.: 理论计算结合实验,揭示非氧化还原Ni3+催化有机物的亲核电氧化大多数有机物电氧化所需的电位远低于高能耗的水氧化。因此,用热力学上有利的有机物电氧化取代水分解OER提供了一种节能的制氢策略,并同时能够生产高附加值化学品或处理工业废水。Ni基电催化剂广泛应用于大规模的工业水电解,并且在有机物的电氧化中也具有高活性。然而,在Ni基催化剂上的水氧化和有机物氧化电化学过程有很大的不同:对于Ni(OH)2电极,水氧化通常发生在Ni2+(OH)2/Ni3+OOH氧化之后,并且它们的电位差足够大以至于可以使用Ni(OH)2/NiOOH作为氧化还原对解耦OER和析氢反应(HER)。这意味着,尽管Ni2+(3d8,t2g6eg2)通过Ni(OH)2→NiOOH+h++e−氧化为Ni3+(3d7,t2g6eg1),但是NiOOH仍然不能像高价Niδ+(δ>3)一样氧化水。相反,与OER过程完全不同,一旦Ni(OH)2被氧化成NiOOH,有机物立即被电解。然而,由于在Ni基催化剂上水和有机物氧化的性质存在较大差异,催化机理尚不清楚,极大地阻碍了高效催化剂的设计和制备。最近,南京大学闫世成课题组使用各种理论和实验技术,清楚地表明有机物在NiOOH上的电氧化不遵循Ni(OH)2/NiOOH氧化还原介导的电子转移机制,而是没有Ni3+物种氧化状态变化的非氧化还原电子转移过程。Ni3+作为亲电电氧化中心,与具有最高占据分子轨道(HOMO)能级(−7.4到−6 eV)的有机物(亲核官能团中亲核原子的双局部软度值(∆sk)为−0.65到−0.15)形成等能转移通道。因此,(HOMO,∆sk)组合判据可以很好地解释为什么Ni3+不能有效地氧化水,但对有机物的电氧化起作用。有机物的快速电氧化动力学可归因于亲核攻击Ni活性位建立能量转移通道,以及有机物HOMO能级电子的化学势足够高,可以驱动电子从成键轨道向催化中心转移。虽然水中O原子的∆sk为−0.607,但水的HOMO能级(−7.99 eV)低于NiOOH的费米能级,触发水氧化电子转移的热力学要求较高。因此,可以清晰地描述完整的电化学电子转移过程:对于电催化氧化反应来说,外加电压首先将活性中心从低价态升高到高价态,此过程可能伴随质子耦合电子转移的相变过程;高价态催化活性中心与反应中间体通过轨道交叠形成成键轨道作为电子转移的能量通道,二者之间电子转移遵循Marcus电荷转移理论,而催化中心则通过高价态活性离子的未占据轨道直接转移电子,此时催化中心并不发生价态变化,通常遵循双/超交换机制转移电子。此外,为了评价Ni3+氧化有机物的实际应用潜力,进一步估算了Ni3+的氧化能力。将NiOOH阳极移至含有有机物的电解液中,Ni3+与有机物发生自发的化学反应,可将NiOOH阳极还原为Ni(OH)2。因此,可以把这个过程分为两个步骤:一个电化学步骤,在电解液中阴极产生H2和氧化阳极形成NiOOH;随后一个自发的化学步骤,通过氧化有机物把阳极还原到初始状态。空间分离的两步法可以在不同的反应室实现制氢和有机物的氧化,从而有利于生产高纯度的产品。Nonredox trivalent nickel catalyzing nucleophilic electrooxidation of organics. Nature Communications, 2023. DOI: 10.1038/s41467-023-43649-6 3. JACS: STM/AFM结合理论计算,证实界面水促进Cu(111)上甲酸去质子化甲酸(FA,HCOOH)是最简单的羧酸,因其具有较高的容量(53.4 g H2/L)和室温下的液体性质,在储氢领域受到越来越多的关注。通常FA的分解包括两个平行的途径,即脱氢(产生CO2和H2)和脱水(产生CO和H2O)。值得注意的是,FA在气相中主要发生脱水反应,但在水相中更易发生脱氢反应,水的存在降低了脱氢反应的反应势垒。目前大多数的研究都是针对少数FA和水分子进行理论计算。为了全面理解反应机理,必须考虑分子间氢键相互作用和分子自组装以及固体表面的质子转移过程。由于FA和水分子都是强氢键的供体和受体,它们氢键相互作用的复杂性可以显著地决定它们的催化反应。但是,到目前为止,水如何参与催化剂表面上FA的分解仍然不清楚,因此需要在原子尺度上对反应过程进行高分辨率表征。基于此,北京师范大学郭静、北京理工大学曹端云和南方科技大学郭庆等系统地研究了水对FA在Cu(111)上的表面化学和反应的影响,并证明需要相对较大的水/FA比率来实现FA的完全去质子化和高效的H2生产。具体而言,研究人员使用STM/AFM结合密度泛函理论(DFT)计算,将界面水对Cu(111)上FA去质子化的促进作用可视化,并在原子水平上洞察了反应机理。结果表明,首先,FA与水在Cu(111)表面共吸附时发生解离,生成H+离子和HCOO−离子;同时,大多数水合质子和HCOO−在Cu(111)上表现出相分离行为,其中Eigen和Zundel阳离子组装成单层六方氢键(H-键)网络,并且双齿HCOO−离子被水溶解并聚集成一维链或二维氢键网络。这种相分离行为对于阻止质子从H+离子向HCOO−的转移和FA的分解是必不可少的。密度泛函理论(DFT)计算表明,水作为酸的强受体(碱),导致了H+离子和HCOO−离子的产生,降低了FA在Cu(111)上的去质子化能垒,其中水通过Grotthuss质子转移机制催化FA的分解。综合以上结果,发现需要大量过量的水来促进FA的完全去质子化,其中额外的水作为溶剂,导致形成水-甲酸盐复合物和离子-水覆盖物,这也可以解释为什么需要过量的水来促进FA的完全分解。此外,由于与Cu基底的相互作用增强,H+离子和双齿HCOO−离子的单独溶剂化是能量上的优先选择。此外,程序升温脱附实验显示,水和FA共吸附在Cu(111)表面时,H2脱附峰强度显著增强,FA脱附降低,进一步表明水对FA脱质子化的促进作用。Visualizing the promoting role of interfacial water in the deprotonation of formic acid on Cu(111). Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c07726 4. JACS: 原子催化剂构型的可逆调整,实现高效氧电还原氧还原反应(ORR)是燃料电池、金属-空气电池等可再生能源装置中必不可少的组成部分。为了推进这些装置的实际应用,设计一种能够有效改善ORR缓慢动力学的电催化剂至关重要。具有M-N-C结构的单原子催化剂作为氧还原反应(ORR)的高效电催化剂引起了人们的广泛关注。值得注意的是,尽管具有相似的M-N-C结构,但是由不同载体(ZIF和石墨相氮g-C3N4)负载的SAC不一定具有相似的催化性能,也就是载体可能导致特征性的金属载体相互作用(MSI),这可能进一步诱导不同的M-N配位构型,并引起不同的催化行为。因此,在设计SAC时必须考虑这种配置依赖性。然而,在ORR过程中,由衍生框架诱导的SACs的动态M-N构型的作用仍然知之甚少。基于此,台湾大学陈浩铭和Jiali Wang等制备了一系列固定在g-C3N4和ZIF载体上的具有可控Cu-N构型的Cu SAC作为模型催化剂,来了解动态结构演变和MSIs对ORR催化行为的影响。研究结果表明,g-C3N4上的Cu SAC表现出对称的Cu-N配位构型,在操作条件下具有可逆的自适应性质,导致其优异的ORR催化活性;相反,ZIF衍生的Cu SAC上的Cu-N构型由于其结构的不对称性,在ORR过程中发生了不可逆的结构变化,其中拉长的Cu-N对在ORR过程中不稳定且会发生断裂(该过程与ORR反应竞争),导致ORR的高过电位。除了配位情况,另一个影响ORR活性增强的因素是相互连接的碳骨架,它可以显著影响电荷转移过程。与具有相同热解温度的裸基底相比,将Cu金属掺入g-C3N4后,C K边XAS的光谱特征保持不变;然而,当Cu固定在ZIF骨架上时,局部碳骨架发生重构,导致C=O键的富集。值得注意的是,与1s到π*跃迁相关的285.5 eV峰的强度在ZIF衍生的Cu SACs中显示出显著的减弱,这种降低可能会降低电荷转移效率,从而影响ORR性能。总的来说,上述发现为Cu-N构型的动态变化与ORR动力学之间的联系提供了新见解,并证明了SAC中Cu-N构型的动态适应对于电催化反应的重要性。Reversibly adapting configuration in atomic catalysts enables efficient oxygen electroreduction. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c10707 5. JACS: 调节催化剂表面Ni3+和OV含量,定向电合成己二酸和环己酮二元羧酸和环酮类化合物,如己二酸(AA)和环己酮(CHN),是化学工业中必不可少的化合物。以水为氧源在温和条件下将木质素衍生的环己醇(CHA)转化为CHN和AA被认为是最具竞争力的策略。其中,Ni基催化剂由于其高活性、丰富性和稳定性,被认为是电氧化研究中最有效的电极材料之一。最近,据报道,用十二烷基磺酸钠(Ni(OH)2-SDS)修饰或掺杂Cu的Ni(OH)2催化剂(Cu-Ni(OH)2)在恒电位(≥1.5 VRHE)下表现出超过80.0%的AA收率。为了达到更高的AA收率(>90%)以满足当前工业生产的要求,通过研究从CHA到AA的整个反应中涉及的每个步骤(即CHA脱氢和CHN氧化)的机理并同时探索它们的匹配过程来开发更先进的电催化剂至关重要。此外,对CHA脱氢反应的深入了解,有助于控制CHA的电化学反应程度,从而获得较高的CHN收率,这对于工业上合成己内酰胺(尼龙-6的反应物)、硝化纤维素和涂料等精细化学品和溶剂具有重要意义。近日,复旦大学唐颐、徐昕和暨南大学高庆生等通过金属基体组分的自溶解、界面生长和电化学活化制备了一系列整体式msig/ea-NiOOH-Ni(OH)2/NF (msig/ea)。光谱表征和理论计算表明,催化剂表面O2−位点可以显著加速CHA中C-H和O-H键的脱氢,而在NiOOH上的OV位点上产生的*OOH物种可以有效提高CHN氧化的活性。通过控制电化学氧化还原活化过程,可以精细地调节表面Ni3+和OV的含量。在优化的活性中心和反应条件下,单步反应(CHA脱氢和CHN氧化)的CHN收率和AA收率分别达到96.5%和93.6%。考虑到最佳电催化剂上CHA转化为CHN和CHN转化为AA的选择性/收率极高,研究人员将CHA脱氢和CHN氧化这两个体系进行了耦合(以CHA为底物)。结果表明,CHA首先在与Ni3+结合的O2−上脱氢最转化为CHN,然后形成的CHN在*OOH上氧化并最终获得了高达92.2%的AA收率,超过了以往所有相关的研究。综上,这项工作深入研究了分步电化学反应的结构-功能关系,为精确设计更先进的工业催化剂以定向电合成高纯度、高附加值的化学品提供了参考。Directional electrosynthesis of adipic acid and cyclohexanone by controlling the active sites on NiOOH. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c05898 6. JACS: 揭示扩展的不对称四配位水网络在HER反应中的主导作用析氢反应(HER)作为一种典型的电化学反应,是理解以水为介质或反应物的各种电化学反应的理论基础的良好模型,对于将液态水转化为高价值氢燃料具有重要意义。因此,准确描述电极/溶液界面处的水取向、氢键环境和结构转化对于揭示控制源自水解离和溶剂环境的HER活性的关键因素至关重要。最近,氢键网络的连接性和无序性被相继确定为HER的主导因素。因此,揭示界面水的功能需要精确识别界面水的精细结构和氢键网络的动态变化以及分子水平上双电层(EDL)中复杂的相互作用,但这仍然是一个挑战。最近,清华大学李景虹和中国科学院长春应化所姜秀娥等结合原位SEIRA光谱电化学、1H核磁共振波谱和MD模拟,揭示了水结构与HER活性之间的关系。研究发现,水分子从孤立到水团簇甚至更大的氢键网络的演化将导致HER活性的逐渐增强。在高负极化电位下,水分子的O-H键会断裂,形成H*中间体和OH−离子;由此产生的电荷被直接与Au膜相互作用的相对无序的非对称四配位水网络传输离开界面,而具有强氢键的对称四配位水网络由于其刚性和远离界面的特点,可能不利于HER过程。这成功区分了与电极相互作用的水和外层水,阐明了不同结构的水在电化学反应中的重要作用,对于理解以水为反应物或介质的电化学反应的基本步骤具有重要意义。此外,通过添加亲水性和疏水性阳离子,发现亲水性Li+离子在短程范围内会促进水的解离,但在长程范围内会破坏水网络的连通性;而疏水性三丁基溴化铵阳离子则极大地促进了界面处形成扩展的不对称四配位网络,有利于OH−离子通过EDL的Volmer步骤传输,从而增强了HER。因此,只有离子-水局域相互作用和氢键网络连接性的协同作用才能主导最佳的水解离和电荷传输,从而获得最高的HER活性。综上,该项工作清楚地揭示了依赖于电位的不对称四配位水网络的连接性受亲水和疏水阳离子的调控,并与HER活性正相关,为揭示水在催化、能源和表面科学中的功能提供了参考。Uncovering the dominant eole of an extended asymmetric four-coordinated water network in the hydrogen evolution reaction. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c08333 7. ACS Catal.: Cu基电催化剂表面羟基化,促进电化学还原CO2电化学CO2还原反应(CO2RR)为CO2直接转化为高附加值化学品和燃料提供了一条可持续的途径。在众多CO2RR产物中,多碳(C2+)产品由于其与C1产品相比具有更高的能量密度和市场价值而受到广泛关注。由于Cu表面上最佳的*CO结合能,其可作为具有最佳C2+产物选择性的CO2RR催化剂。迄今为止,在高性能铜基电催化剂的结构设计和精确制备方面取得了重大进展。并且,许多工作已经开始着手研究与电极界面微环境调控相关的一些关键因素和策略,如催化剂表面的分子工程、电解质设计和电解池构型等,这促使人们在设计和制备催化剂以引导对目标产物的选择性时应始终考虑催化剂周围的反应微环境。目前,研究较多的策略之一是利用浓KOH将CO2地转化为C2+产物,但阴极上碳酸盐的生成消耗了大部分输入的CO2。因此,寻求一种新的策略来改善电极局部环境,从而消除或降低CO2RR选择性对本体OH−浓度的依赖至关重要。基于此,华东理工大学李春忠、李会会和日本东北大学李昊等通过羟基功能化的表面策略(即在Cu2O催化剂上覆盖上富含羟基的分子)来实现分子表面修饰以增强C2+产物的形成。电化学实验和原位表征证实了在CO2RR反应过程中,催化剂表面附近稳定存在的羟基物种能够有效地将吸附的*CO转化为C2+产物。在流动池中,最优的0D-Cu-800催化剂上C2+产物的法拉第效率为81.5%,部分流电流密度为285 mA cm−2,以及阴极能量效率为43.1%;利用阳离子交换膜电极组件装置,研究人员证明了在平均电流密度为151 mA cm−2的条件下,0D-Cu-800上能够连续稳定生产C2H4超过100小时。理论计算表明,富含羟基的分子如葡萄糖酸可以导致Cu位点的电子损失,这有助于改善CO与Cu之间的电子转移,进而促进*CO的吸附和C-C耦合,提高了电化学CO2RR对C2+产物的选择性。总的来说,该项工作证明了表面修饰对于设计稳定的反应微环境的重要性,这为未来设计高效的电催化剂以提高CO2RR或其他电化学反应活性提供了指导。Boosting electrochemical CO2 reduction via surface hydroxylation over Cu-based electrocatalysts. ACS Catalysis, 2023. DOI: 10.1021/acscatal.3c02454 8. Adv. Sci.: F迁移耦合双金属中心,促进材料表面重构以提升OER活性电化学水分解是一种生产高纯度氢气的有效途径。然而,在阳极半反应中,由于多步质子/电子耦合,析氧反应(OER)比阴极析氢反应(HER)需要更大的过电位,这阻碍了能量的有效转化。其中,RuO2和IrO2等贵金属催化剂能够显著降低OER的过电位,但其昂贵的价格和稀缺的储量严重限制了它们的大规模工业应用。在众多贵金属催化剂替代品中,过渡金属基电催化剂由于其价廉、储量丰富等优点而得到了广泛的研究。其中,过渡金属氟化物中的F具有最大的电负性,可以将金属中心的价态推向最高,这有助于辅助催化剂表面重构、形成非晶结构等。因此,氟化物可能是OER的有效催化剂,但目前文献中很少有关其的报道。近日,武汉大学赵苹苹和程功臻等设计并制备了一种结晶性良好的NiCo双金属氟化物(Ni0.42Co0.58F2-G),并研究了其在电化学过程中的重构现象。具体而言,研究人员首先采用固液交换法合成了具有较大比表面积的NiCo基纳米阵列A-Ni-MeIM-Co-0.01前体,再通过一步气相氟化法将其转化为具有较高电化学活性表面积的NiCo双金属氟化物Ni0.42Co0.58F2-G。与传统的液相合成方法相比,气相氟化法生产的氟化物具有更高的结晶度;同时,双金属中心电子结构的调控在电化学过程中促进了高效的表面重构以及重构过程中F的迁移导致更多电化学活性位点的暴露。这不仅使Ni0.42Co0.58F2-G具有优异的电化学性能,而且保证了其电化学稳定性。电化学性能测试结果显示,所制备的Ni0.42Co0.58F2-G催化剂在10 mA cm−2电流密度下的OER过电位仅为313 mV,Tafel斜率为42.5 mV dec−1;并且,该催化剂在10000次CV循环后活性仍保持稳定,表明其具有优异的稳定性。此外,理论计算表明,双金属中心的构建可以诱导电荷重新分布并产生更高价金属位点,从而优化关键反应中间体的吸附,降低反应能垒,显著提高OER活性。综上,该项工作详细研究了F和双金属中心在提升催化剂OER活性中的作用,这对于设计和制备高效的电化学表面重构电催化剂具有一定的指导意义。Surface reconstruction facilitated by fluorine migration and bimetallic center in NiCo bimetallic fluoride toward oxygen evolution reaction. Advanced Science, 2023. DOI: 10.1002/advs.202306758【做计算 找华算】华算科技专注DFT代算服务、正版商业软件版权、全职海归计算团队,10000+成功案例!客户成果发表在Nature、Nature Catalysis、JACS、Angew.、AM、AEM、AFM等顶刊,好评如潮,专业靠谱!添加下方微信好友,立即咨询: